library(rpact)
packageVersion("rpact")Planning and Analyzing a Group-Sequential Multi-Arm Multi-Stage Design with Binary Endpoint using rpact
Planning
Rates
Multi-arm
Analysis
This document provides an example of implementing, simulating and analyzing multi-arm-multi-stage (MAMS) designs for testing rates with rpact with special regards to futility bound determination, treatment arm selection and generic data analysis. After exemplarily using the binary endpoint analysis module from rpact, an illustrative landmark analysis (comparison of empirical survival probabilities at specific time point) using Greenwoods standard error estimation is to be performed. Since rpact itself does not directly support this type of analysis, another packages’ functionality needs to be utilized to perform the survival probability and standard error estimation to eventually use the estimates as input for a hypothetical continuous endpoint dataset which subsequently is to be analyzed as such.
Introduction
This document provides an exemplary implementation of multi-arm-multi-stage (MAMS) design with a binary endpoint using rpact. Concretely, the vignette consists of considering design implementation with respect to futility bounds on treatment effect scale, sample size calculations and simulations given different treatment selection approaches. Furthermore, analysis using closed testing will be performed on generic binary and survival data.
Note that rpact itself does not support landmark analysis, i.e. comparison of survival probabilities at a fixed point in time, using the Greenwoods standard error (SE) formula. Thus, the first two analyses are based on the empirical event rates only. The R packages gestate and survival are then utilized to briefly show how one could merge the packages to perform the actually intended analysis using boundaries obtained by rpact and test statistics obtained by survival probabilities and standard errors estimated using gestate and survival.
For methodological and theoretical background, refer to “Group Sequential and Confirmatory Adaptive Designs in Clinical Trials” by Gernot Wassmer and Werner Brannath.
Before starting, load the rpact package and make sure the version of the package is at least 3.1.0:
[1] '4.4.0.9305'Implementation of design
Consider we are interested in implementing a group sequential design with \(3\) stages, \(3\) treatment arms (\(2\) active, \(1\) control), \(2\) treatment arm comparisons (each active arm vs. the control arm), a binary endpoint, with a global one-sided \(\alpha=0.025\), and the power for each treatment arm comparison set to \(80\%\), hence \(1-\beta=0.8\). Additionally, let the critical boundaries be calculated using alpha-spending O’Brien & Fleming approach and for planning purposes, suppose equally distributed information rates. Further, non-binding futility bounds are to be set in the following way:
Considering the \(2\) active treatment arms, the goal of both arms is to confirm significant reduction in event (e.g., disease, death) rates as compared to the control arm. Thus, a treatment should be considered futile if there is only a small reduction, no reduction or even an increase in the event rates seen in the data. This can be implemented in a study design in which a treatment arm is considered futile after the first stage if the relative reduction does not exceed \(5\)% (i.e., a rate ratio \(\geq 0.95\)) and after the second stage in case the data indicates a relative reduction of \(10\)% or less (i.e., a rate ratio \(\geq 0.9\)). As rpact primarily uses futility bounds specified on the \(z\)-scale, one needs to determine the \(z\)-scale values that correspond to the values listed above. To achieve these values, we may use the fact that the reverse operation, i.e., the calculation of futility bounds on the treatment effect scale given futility bounds specified on the \(z\)-scale, is a part of the routine output of sample size calculation in rpact (given certain treatment effect assumptions). We may thus either heuristically try out various options for futility bounds on the \(z\)-scale and attempt to get as close as possible to the desired futility bounds on the treatment effect scale or implement a search algorithm for higher precision. For simplicity, we assume that the stage-wise futility bounds are independent.
For the design example mentioned above, this can be achieved as follows:
First, one needs to initialize the design and use more or less arbitrary futility bounds on the \(z\)-scale as input:
# first and second stage futility on z-scale
fut1 <- 0.16
fut2 <- 0.39
d_fut <- getDesignGroupSequential(
kMax = 3,
alpha = 0.025,
beta = 0.2,
sided = 1,
typeOfDesign = "asOF",
informationRates = c(1/3, 2/3, 1),
futilityBounds = c(fut1, fut2),
bindingFutility = FALSE
)Note that the specified futility bounds on the \(z\)-scale are positive, even though they are intended to later correspond to a reduced risk ratio.
Now, as mentioned, the sample size calculation output provides information about the futility boundaries on the treatment effect scale. Therefore, after assuming certain treatment effects, one needs to perform sample size calculation and extract the treatment effect futility bounds from the respective output:
c_assum <- 0.1 # assumed rate in control
effect_assum <- 0.5 # relative reduction that is to be detected with probability of 0.8
# rates indicate binary endpoint
ssc_fut <- getSampleSizeRates(
design = d_fut,
riskRatio = TRUE,
pi1 = c_assum * (1 - effect_assum),
pi2 = c_assum
)
ssc_fut$futilityBoundsEffectScale [,1]
[1,] 0.9464954
[2,] 0.9085874The values printed out above are now the futility bounds on the treatment effect scale which correspond with the values fut1 and fut2 from above. Since \(0.9465 < 0.95\) and \(0.9086 > 0.9\), the input value fut1=\(0.16\) seems to be slightly too large while fut2=\(0.39\) seems to be slightly too small (since, in our example, a larger \(z\)-value corresponds to a smaller risk ratio). This indicates how to adjust the input values such that one comes closer to the true corresponding values.
Now, using a one-dimensional search algorithm for each stage or by simply trying different values, one achieves the futility bounds on the \(z\)-scale summarized in the following table:
| First stage | Second stage | |
|---|---|---|
| Orig. treatment effect scale | 0.950 | 0.900 |
| Approx. z-Value for given t-Value | 0.149 | 0.414 |
| Corresponding t-scale Value | 0.950 | 0.900 |
| Diff. between Approx. and Input | 0.000 | 0.000 |
In the table above, the Orig. treatment effect scale is the intended futility bound on treatment effect scale, Approx. \(z\)-value denotes the corresponding \(z\)-scale value approximation and Corresponding \(t\)-scale Value the actual treatment-effect-scale value using the calculated approx. \(z\)-values as input.
One can see that the first stage futility bound on \(z\)-scale should approximately be chosen as \(0.14915\) and the second stage \(z\)-scale futility bound as \(0.41381\), resulting in the desired futility bounds on the treatment effect scale of approximately \(0.95\) in the first and \(0.9\) in the second stage, respectively. Note that, for simplicity, these futility bounds were identified separately for the first and second stage and under the assumption that, at the respectively other stage, no futility stopping is performed. Therefore, when both futility bounds are used at the same time, small numerical deviations from the target values may occur. In the next chapter on sample size calculation, the respective outputs validate that these \(z\)-scale futility bounds are reasonable choices.
Now that (approximate) futility bounds on the \(z\)-scale are known, the design specified above can be entirely initialized:
# GSD with futility bounds according to above calculations
d <- getDesignGroupSequential(
kMax = 3,
alpha = 0.025,
beta = 0.2,
sided = 1,
typeOfDesign = "asOF",
informationRates = c(1/3, 2/3, 1),
futilityBounds = c(0.149145, 0.41381),
bindingFutility = FALSE
)
d |> summary()Sequential analysis with a maximum of 3 looks (group sequential design)
O’Brien & Fleming type alpha spending design, non-binding futility, one-sided overall significance level 2.5%, power 80%, undefined endpoint, inflation factor 1.0833, ASN H1 0.8652, ASN H01 0.843, ASN H0 0.6133.
| Stage | 1 | 2 | 3 |
|---|---|---|---|
| Planned information rate | 33.3% | 66.7% | 100% |
| Cumulative alpha spent | 0.0001 | 0.0060 | 0.0250 |
| Stage levels (one-sided) | 0.0001 | 0.0060 | 0.0231 |
| Efficacy boundary (z-value scale) | 3.710 | 2.511 | 1.993 |
| Futility boundary (z-value scale) | 0.149 | 0.414 | |
| Cumulative power | 0.0213 | 0.4471 | 0.8000 |
| Futility probabilities under H1 | 0.062 | 0.011 |
Printing a summary of the defined object gives a nice overview of all relevant design parameters. Note that the adjusted \(\alpha\) of the last stage (\(=0.0231\)) is only slightly lower than the predefined global \(\alpha\) (of \(0.025\)), corresponding to the final critical value of \(1.993\) being only slightly larger than \(1.96\), which is due to alpha-spending O’Brien Fleming adjustment. In addition, an overview of the input parameters is provided.
Sample size calculation
When it comes to sample size calculations for designs with binary endpoints, rpact provides the function getSampleSizeRates(). It should be noted upfront that the default sample size calculation in rpact only allows for one- or two-group comparisons.
The sample size calculation code applicable here has already been used to determine the \(z\)-scale futility boundaries whenever futility boundaries are only given on the treatment effect scale. However, this sections’ purpose now is to provide some more detail on sample size calculations in MAMS-designs with binary endpoint.
Suppose that, in a two-group setting, we define \(H_0: \pi_1-\pi_2 \geq 0\), with \(\pi_1\) representing the assumed event rate in the treatment group and \(\pi_2\) being the assumed event rate in the reference/control group. Again, let the parameter setting be given as above and assume an expected relative reduction of event occurrence of \(50\%\) given \(\pi_2 = 0.1\) (hence \(\pi_1 = 0.1\cdot0.5=0.05\)). Since we are interested in comparing risks via their ratio, we set riskRatio = TRUE which particularly results in testing \(H_0: \frac{\pi_1}{\pi_2} \geq 1\) against \(H_1:\frac{\pi_1}{\pi_2} < 1\). The corresponding sample sizes of the trial design defined above can be calculated using the commands:
c_rate <- 0.1 # assumed rate in control
effect <- 0.5 # relative reduction that is to be detected with probability of 0.8
# rates indicate binary endpoint
d_sample <- getSampleSizeRates(
design = d,
riskRatio = TRUE,
pi1 = c_rate * (1 - effect),
pi2 = c_rate
)
d_sample |> summary()Sample size calculation for a binary endpoint
Sequential analysis with a maximum of 3 looks (group sequential design), one-sided overall significance level 2.5%, power 80%. The results were calculated for a two-sample test for rates (normal approximation), H0: pi(1) / pi(2) = 1, H1: pi(1) = 0.05, control rate pi(2) = 0.1.
| Stage | 1 | 2 | 3 |
|---|---|---|---|
| Planned information rate | 33.3% | 66.7% | 100% |
| Cumulative alpha spent | 0.0001 | 0.0060 | 0.0250 |
| Stage levels (one-sided) | 0.0001 | 0.0060 | 0.0231 |
| Efficacy boundary (z-value scale) | 3.710 | 2.511 | 1.993 |
| Futility boundary (z-value scale) | 0.149 | 0.414 | |
| Efficacy boundary (t) | 0.061 | 0.476 | 0.643 |
| Futility boundary (t) | 0.950 | 0.903 | |
| Cumulative power | 0.0213 | 0.4471 | 0.8000 |
| Number of subjects | 313.8 | 627.5 | 941.3 |
| Expected number of subjects under H1 | 751.7 | ||
| Overall exit probability (under H0) | 0.5594 | 0.1828 | |
| Overall exit probability (under H1) | 0.0838 | 0.4366 | |
| Exit probability for efficacy (under H0) | 0.0001 | 0.0059 | |
| Exit probability for efficacy (under H1) | 0.0213 | 0.4258 | |
| Exit probability for futility (under H0) | 0.5593 | 0.1769 | |
| Exit probability for futility (under H1) | 0.0625 | 0.0108 |
Legend:
- (t): treatment effect scale
The variable Futility boundary (t) represents the futility bounds transferred to treatment effect scale. The futility boundaries calculated above correspond to a rate ratio of \(0.950\) at the first stage (i.e., a relative reduction of \(5\%\)) and a rate ratio of \(0.903\) at the second stage (i.e., a relative reduction of approx. \(10\%\)). As mentioned above, the slight differences to the targeted risk ratios are due to the fact that the futility bounds are calculated independently. However, these differences are small and only occur at the third digit. (The keen-eyed reader may have noticed that the first-stage futility boundary is also not perfectly exact. This is because the sample sizes, which influence the transformation from \(z\)- to treatment effect scale, change slightly when the second-stage futility boundary is introduced.)
Another important information provided by the output is that to achieve the desired study characteristics, such as keeping the \(\alpha\)-error controlled at \(0.025\) while obtaining a power of \(1-\beta = 0.8\), approx. \(\frac{314}{2}=157\) study subjects are needed per arm per stage given equal information rates. For a design with \(3\) treatment arms, of which \(2\) are active treatment arms and \(1\) is a control arm, one may choose to simply extrapolate the (maximum) number of subjects by \(3\cdot 3\cdot 157 = 1413\), i.e., multiplying the sample size per arm and stage by the \(3\) arms and \(3\) stages.
It should be noted here that this is just an approximation of the sample size needed to achieve a power of \(1-\beta=0.8\), while power here is defined as the probability of successfully detecting the assumed effect. Simulations in following chapters will indicate that this rather rough approximate sample size is actually rather large since the studies appear to be overpowered in simulations, while the term power has a different meaning here: when considering simulations of studies with multiple active treatment arms, power is referred to as the probability to claim success of at least one active treatment arm in the study.
The row Exit probability for futility indicates the rather low probability of early stopping due to futility (Stage 1: \(0.0625\), Stage 2: \(0.108\)), while this depends on the assumed treatment effects and the defined boundaries. Efficacy boundary (t), just as Efficacy boundary (z-value scale) shows that a drastic decrease in events needs to be detected in the treatment group in order to reject \(H_0\) in the first stage (rate ratio of \(0.061\), i.e. relative reduction of \(1-0.061=0.939\)) which, again reflects the “conservatism” of alpha-spending O’Brien Fleming approach in early stages, however resulting in rather liberal and monotonically decreasing boundaries along the stages.
# boundary plots
par(mar = c(4, 4, .1, .1))
plot(d_sample, main = paste0(
"Boundaries against stage-wise ",
"cumul. sample sizes - 1"
), type = 1)
plot(d_sample, main = paste0(
"Boundaries against stage-wise ",
"cumul. sample sizes - 2"
), type = 2)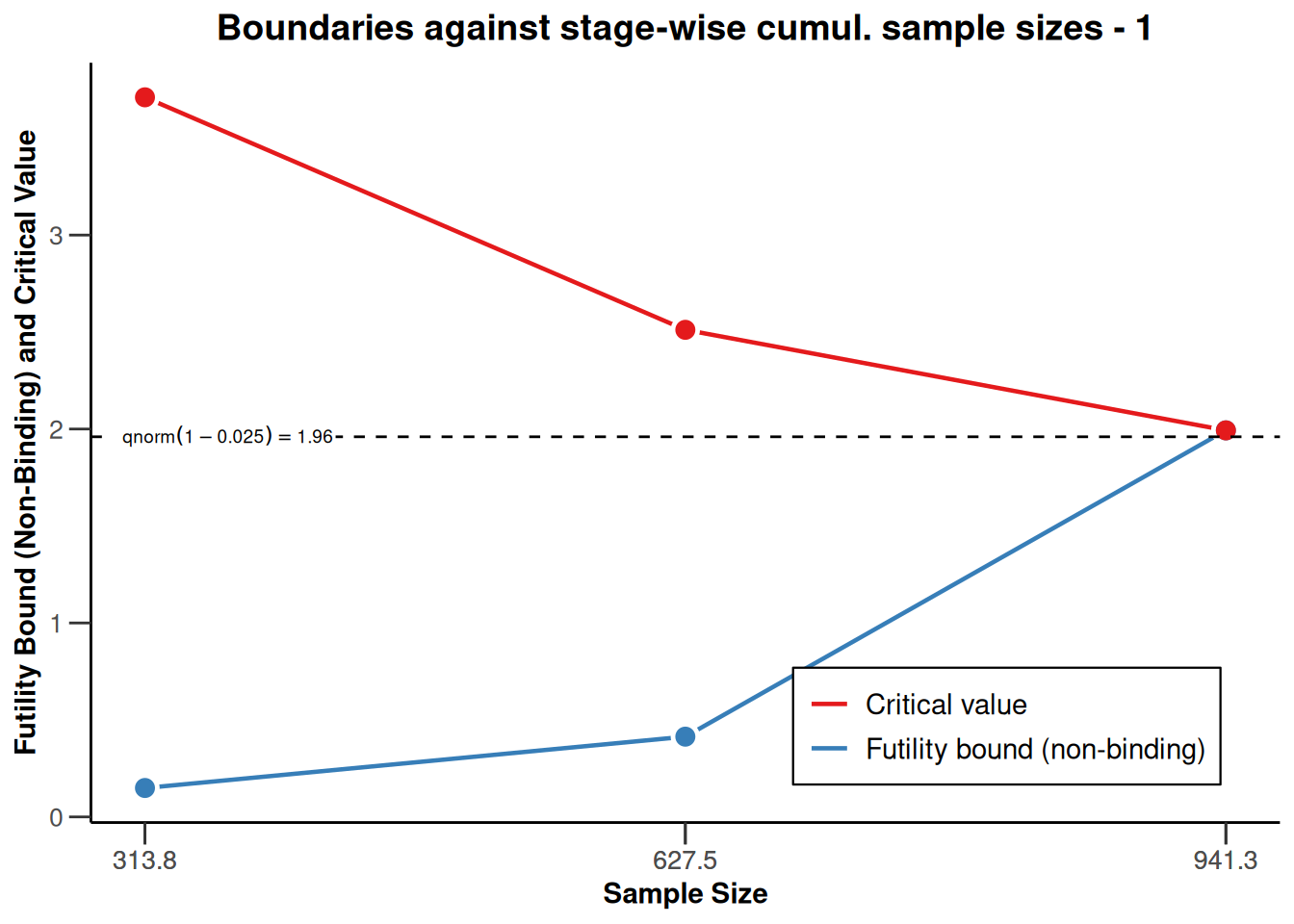
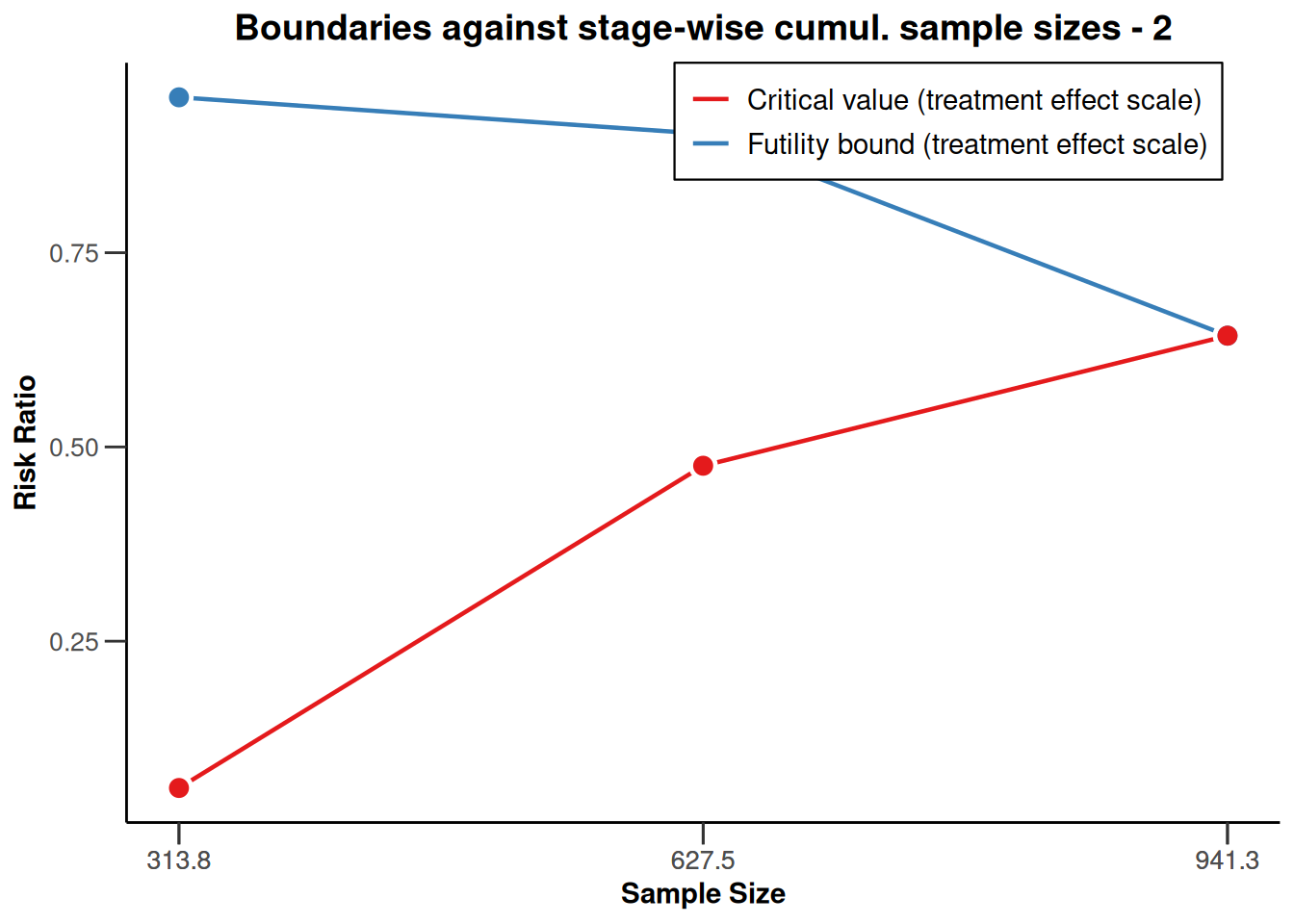
Plotting the cumulative sample size against the applicable boundaries is a way of nicely visualizing these important study characteristics. The upper plot displays the stage-wise (\(z\)-scale) boundaries of the trial design. The red line marks the efficacy bounds which need to be crossed to obtain statistical significance at each stage, the blue line represents the futility bounds. The dashed constant line represents the critical value of a fixed study design with one-sided testing and \(\alpha=0.025\), i.e., with a critical value of \(\Phi^{-1}(1-\alpha) \approx 1.96\) (\(\Phi^{-1}\) denoting the quantile function of the standard normal cumulative distribution function). The lower plot contains the same information, however the y-axis is now rescaled to represent the treatment effect scale. The color-coding of efficacy (red) and futility (blue) bounds is the same as in the plot above. Note that the plot again indicates that low risk ratio values are desirable.
Study simulations
Performing simulations prior to conducting a study is often reasonable since they allow for evaluating characteristics, such as power (please refer to definition above), assuming different scenarios or constellations of treatment effects. Thus, simulations allow for a more holistic view on how the planned study could go in case the assumptions are (approximately) true.
Simulation of the study design will be done using the function getSimulationMultiArmRates(), which needs an Inverse Normal Design as input, defined with the same arguments as the design used above:
# design as above, just as inverse normal
d_IN <- getDesignInverseNormal(
kMax = 3,
alpha = 0.025,
beta = 0.2,
sided = 1,
typeOfDesign = "asOF",
informationRates = c(1/3, 2/3, 1),
futilityBounds = c(0.149145, 0.41381),
bindingFutility = FALSE
)To perform study simulations for, e.g., the evaluation of power or probability-of-success, effect rates in the \(2\) active treatment arms must be assumed and specified in a matrix. For binary data, the effectMatrix refers to the event rate assumed in each arm. In addition, we set the (maximum) number of iterations for the simulations in advance.
# set number of iterations to be used in simulation
maxNumberOfIterations <- 10000
# specify the scenarios, nrow: number of scenarios, ncol: number of treatment arms
effectMatrix <- matrix(c(0.100, 0.100, 0.05, 0.05, 0.055, 0.045),
byrow = TRUE, nrow = 3, ncol = 2
)
# first column: first treatment arm, second column: second treatment arm
show(effectMatrix) [,1] [,2]
[1,] 0.100 0.100
[2,] 0.050 0.050
[3,] 0.055 0.045Each row of the effect matrix specifies a scenario to be considered in the simulation. Considering a design with \(2\) active treatment arms, both to be compared against a common control arm, the effect matrix contains the event rates of the first active arm in the first column and the event rates of the second active arm in the second column, respectively. The event rate of the control group is not a part of the effect matrix and is later provided directly to getSimulationMultiArmRates().
Now that trial design and effect matrix have been derived, the simulations can be performed. In order to appropriately model the specific situation of a trial, a wide range of options is available for the simulation function getSimulationMultiArmRates(). Choosing typeOfShape as "userDefined" determines that the effect matrix specified above should be used for the simulations. For more specific applications, in which the different treatment arms correspond to different dose levels, typeOfShape may be used to specify a dose-response relationship (e.g., typeOfShape = "linear" for a linear dose-response relationship). Depending on the chosen type of shape, additional arguments may need to be specified. For details, refer to the function documentation (?getSimlulationMultiArmRates). directionUpper refers to the direction of the alternative and is set to FALSE, since, in our example, the alternative hypothesis is directed towards lower values, i.e., a reduction of event rates is considered beneficial to the subjects. The intersection test to be performed is Simes (intersectionTest = "Simes"), with other options being, e.g., "Bonferroni" or "Dunnett" (the default setting). typeOfSelection is initially set to "rBest" with rValue = 2, which means that at each stage, the \(2\) best treatment arms should be carried forward. successCriterion = "all" means that, in order to stop the trial early due to efficacy, both active treatment arms need to perform significantly better than the control arm at an interim analysis. Another option would be to set successCriterion = "atLeastOne" to declare significant testing of one treatment arm to be sufficient for study success at interim. The vector plannedSubjects contains the stage-wise cumulative sample sizes of the (active) arms. Since, in our example, the control arm should be of the same size as the active arms, we set allocationRatioPlanned = 1.
# first simulation
simulation <- getSimulationMultiArmRates(
design = d_IN,
activeArms = 2,
effectMatrix = effectMatrix,
typeOfShape = "userDefined",
piControl = 0.1,
intersectionTest = "Simes",
directionUpper = FALSE,
typeOfSelection = "rBest",
rValue = 2,
effectMeasure = "testStatistic",
successCriterion = "all",
plannedSubjects = c(157, 314, 471),
allocationRatioPlanned = 1,
maxNumberOfIterations = maxNumberOfIterations,
seed = 145873,
showStatistics = TRUE
)
simulation |> summary()Simulation of a binary endpoint (multi-arm design)
Sequential analysis with a maximum of 3 looks (inverse normal combination test design), one-sided overall significance level 2.5%. The results were simulated for a multi-arm comparisons for rates (2 treatments vs. control), H0: pi(i) - pi(control) = 0, power directed towards smaller values, H1: pi_max as specified, control rate pi(control) = 0.1, planned cumulative sample size = c(157, 314, 471), effect shape = user defined, intersection test = Simes, selection = r best, r = 2, effect measure based on test statistic, success criterion: all, simulation runs = 10000, seed = 145873.
| Stage | 1 | 2 | 3 |
|---|---|---|---|
| Fixed weight | 0.577 | 0.577 | 0.577 |
| Cumulative alpha spent | 0.0001 | 0.0060 | 0.0250 |
| Stage levels (one-sided) | 0.0001 | 0.0060 | 0.0231 |
| Efficacy boundary (z-value scale) | 3.710 | 2.511 | 1.993 |
| Futility boundary (z-value scale) | 0.149 | 0.414 | |
| Reject at least one [1] | 0.0181 | ||
| Reject at least one [2] | 0.8550 | ||
| Reject at least one [3] | 0.8695 | ||
| Rejected arms per stage [1] | |||
| Treatment arm 1 | 0.0001 | 0.0031 | 0.0087 |
| Treatment arm 2 | 0 | 0.0024 | 0.0079 |
| Rejected arms per stage [2] | |||
| Treatment arm 1 | 0.0086 | 0.3839 | 0.3954 |
| Treatment arm 2 | 0.0080 | 0.3796 | 0.3996 |
| Rejected arms per stage [3] | |||
| Treatment arm 1 | 0.0043 | 0.3090 | 0.3968 |
| Treatment arm 2 | 0.0145 | 0.4650 | 0.3686 |
| Success per stage [1] | 0 | 0.0007 | 0.0034 |
| Success per stage [2] | 0.0029 | 0.2735 | 0.4437 |
| Success per stage [3] | 0.0014 | 0.2645 | 0.4228 |
| Stage-wise number of subjects [1] | |||
| Treatment arm 1 | 157.0 | 157.0 | 157.0 |
| Treatment arm 2 | 157.0 | 157.0 | 157.0 |
| Control arm | 157.0 | 157.0 | 157.0 |
| Stage-wise number of subjects [2] | |||
| Treatment arm 1 | 157.0 | 157.0 | 157.0 |
| Treatment arm 2 | 157.0 | 157.0 | 157.0 |
| Control arm | 157.0 | 157.0 | 157.0 |
| Stage-wise number of subjects [3] | |||
| Treatment arm 1 | 157.0 | 157.0 | 157.0 |
| Treatment arm 2 | 157.0 | 157.0 | 157.0 |
| Control arm | 157.0 | 157.0 | 157.0 |
| Expected number of subjects under H1 [1] | 776.2 | ||
| Expected number of subjects under H1 [2] | 1232.8 | ||
| Expected number of subjects under H1 [3] | 1243.7 | ||
| Overall exit probability [1] | 0.5797 | 0.1927 | |
| Overall exit probability [2] | 0.0511 | 0.2803 | |
| Overall exit probability [3] | 0.0443 | 0.2709 | |
| Selected arms [1] | |||
| Treatment arm 1 | 1.0000 | 0.4203 | 0.2276 |
| Treatment arm 2 | 1.0000 | 0.4203 | 0.2276 |
| Selected arms [2] | |||
| Treatment arm 1 | 1.0000 | 0.9489 | 0.6686 |
| Treatment arm 2 | 1.0000 | 0.9489 | 0.6686 |
| Selected arms [3] | |||
| Treatment arm 1 | 1.0000 | 0.9557 | 0.6848 |
| Treatment arm 2 | 1.0000 | 0.9557 | 0.6848 |
| Number of active arms [1] | 2.000 | 2.000 | 2.000 |
| Number of active arms [2] | 2.000 | 2.000 | 2.000 |
| Number of active arms [3] | 2.000 | 2.000 | 2.000 |
| Conditional power (achieved) [1] | 0.0602 | 0.1379 | |
| Conditional power (achieved) [2] | 0.4254 | 0.6771 | |
| Conditional power (achieved) [3] | 0.4290 | 0.6911 | |
| Exit probability for futility [1] | 0.5797 | 0.1920 | |
| Exit probability for futility [2] | 0.0482 | 0.0068 | |
| Exit probability for futility [3] | 0.0429 | 0.0064 |
Legend:
- (i): treatment arm i
- [j]: effect matrix row j (situation to consider)
In this output, the different input situations, which correspond to the rows of the effect matrix, are indicated through \([\,j\,]\) with \(j=1,2,3\).
Under \(H_0\), with assumed treatment effects equal to the assumed effect in the control group (scenario [1]), the probability of rejecting \(H_0\) for at least one treatment arm, i.e., committing a type I error, is controlled at the pre-specified \(\alpha\)-level (Reject at least one [1] \(= 0.0181\leq 0.025\)). For scenarios [2] and [3], which represent parameter constellations under the alternative, the rejection probabilities are high (Reject at least one [2] \(= 0.8550\) and Reject at least one [3] \(= 0.8695\)). In addition, the stage-wise number of subjects are displayed and the probabilities for the arms to be selected at the individual stages are provided for each scenario in the effect matrix. Note that the selection probabilities are the same for the two arms, since rValue = 2 means that, unless the trial is terminated at interim, the two best treatment arms (i.e., all arms in this case) are selected moving forward.
Furthermore, the expected sample size under the provided event rates (Expected number of subjects under H1 [j]), the stage-wise probabilities of a futility stop (Exit probability for futility [j]) as well as the stage-wise conditional power (Conditional power (achieved) [j]), defined as the probability of reaching a statistically significant result given the data observed so far, are listed for each scenario. Note that, due to the relatively high probability of a futility stop in scenario [1] (first stage: \(0.5797\), second stage: \(0.1920\)), the corresponding expected number of subjects is approximately \(776.2\) and thus considerably smaller than the expected sample sizes in the other scenarios.
Stage-wise treatment arm selection
Now, suppose that the underlying treatment arm selection scheme is different and not straightforwardly covered by the other available and pre-defined options in rpact (i.e. "best", "rBest", "epsilon", "all"). In such cases, a user-defined treatment arm selection function can be specified to getSimulationMultiArmRates(). To use an individual selection function, the user must specify typeOfSelection = "userDefined" and provide a selection function in the selectArmsFunction argument. As an example, assume that, at each stage, we only wish to select those treatment arms, for which the observed effect does not fall below the futility bound. Continuing the above example, this means that if the relative rate reduction in a treatment arm does not exceed 5% at the first stage or does not exceed 10% at the second stage, the arm should be deselected at the respective stage.
After the first stage, there are thus six possible analysis results:
- Both active Treatment Arms significant
- 1 active Treatment Arm significant, 1 active Treatment Arm non-significant
- 1 active Treatment Arm significant, 1 active Treatment Arm futile
- Both active Treatment Arms non-significant
- 1 active Treatment Arm non-significant, 1 active Treatment Arm futile
- Both active Treatment Arms futile
Below, we illustrate the treatment arm selection scheme which results from using typeOfSelection = "rBest" with rValue = 2 as above, where the numbers in the circles correspond to the six possible analysis results scenarios.
Selection scheme with rBest, rValue=2
This first diagram graphically represent how treatment selection proceeds choosing typeOfSelection=rBest with rValue=2 in case of the study design previously defined. Early study success can be obtained whenever both treatment arms can be tested significantly at interim and a continuation of the study with only the non-significant (meaning neither stopped for efficacy nor for futility) active treatment arm is to be done whenever the other active treatment arm is discontinued early due to efficacy. If neither of the arms can be tested significantly at interim, the study continues with both treatment arms as even arms crossing the futility bounds are to be carried forward using rBest, rValue=2.
User-defined selection scheme
The second diagram illustrates the active treatment arm selection that is to be implemented through the user-defined selection function. An efficacy stop occurs if both active treatment arms are tested significantly at interim. If only one of the active treatment arms is tested significantly at interim and the other arm is not significant (but also not futile), the trial is continued with the non-significant arm only. The study also terminates early with success of one arm if one active arm is futile while the other can be deemed superior to control. In case both arms are non-significant or one arm is non-significant while the other is futile, the trial is continued with the non-futile arms (or arm) only. Lastly, a futility stop can also happen at interim.
Since futility bounds were already defined in the design specification, one may be led to believe that the selection rule is only an add-on to the futility bound. However, this is not true since specifying a selection rule overwrites the futility bound as the continuation criterion for treatment arms. Thus, even if futility bounds are pre-specified, to implement stopping due to futility in the application of getSimulationMultiArmRates() (and getSimulationMultiArmMeans(), getSimulationMultiArmSurvival()), one could use these as input in the customized selection function. In this case, having the previously defined futility bounds of \(0.149145\) at the first interim and \(0.413808\) at the second respectively, there is a need to individualize the selection scheme for the different stages. It is important to note here that effectMeasure = "testStatistic" needs to be set as the futility bounds have intentionally been transformed to and calculated on the \(z\)-scale. Now, rpact enables implementation of this selection scheme by allowing stage as an argument in the selection function, in addition to effectVector:
# first row: first stage futility bounds, second row: second stage futility bound
futility_bounds <- matrix(c(d_IN$futilityBounds, d_IN$futilityBounds), nrow = 2)
# selection function
selection <- function(effectVector, stage) {
# if stage==1, compare to first stage fut. bounds,
# if stage==2, compare to second stage fut. bounds
selectedArms <- switch(stage,
(effectVector >= futility_bounds[1, ]),
(effectVector >= futility_bounds[2, ])
)
return(selectedArms)
}
simulation <- getSimulationMultiArmRates(
design = d_IN,
activeArms = 2,
effectMatrix = effectMatrix,
typeOfShape = "userDefined",
piControl = 0.1,
intersectionTest = "Simes",
directionUpper = FALSE,
typeOfSelection = "userDefined",
selectArmsFunction = selection,
effectMeasure = "testStatistic",
successCriterion = "all",
plannedSubjects = c(157, 314, 471),
allocationRatioPlanned = 1,
maxNumberOfIterations = maxNumberOfIterations,
seed = 145873,
showStatistics = TRUE
)
simulation |> summary()Simulation of a binary endpoint (multi-arm design)
Sequential analysis with a maximum of 3 looks (inverse normal combination test design), one-sided overall significance level 2.5%. The results were simulated for a multi-arm comparisons for rates (2 treatments vs. control), H0: pi(i) - pi(control) = 0, power directed towards smaller values, H1: pi_max as specified, control rate pi(control) = 0.1, planned cumulative sample size = c(157, 314, 471), effect shape = user defined, intersection test = Simes, selection = user defined, effect measure based on test statistic, success criterion: all, simulation runs = 10000, seed = 145873.
| Stage | 1 | 2 | 3 |
|---|---|---|---|
| Fixed weight | 0.577 | 0.577 | 0.577 |
| Cumulative alpha spent | 0.0001 | 0.0060 | 0.0250 |
| Stage levels (one-sided) | 0.0001 | 0.0060 | 0.0231 |
| Efficacy boundary (z-value scale) | 3.710 | 2.511 | 1.993 |
| Futility boundary (z-value scale) | 0.149 | 0.414 | |
| Reject at least one [1] | 0.0181 | ||
| Reject at least one [2] | 0.8304 | ||
| Reject at least one [3] | 0.8419 | ||
| Rejected arms per stage [1] | |||
| Treatment arm 1 | 0.0002 | 0.0024 | 0.0072 |
| Treatment arm 2 | 0 | 0.0030 | 0.0079 |
| Rejected arms per stage [2] | |||
| Treatment arm 1 | 0.0067 | 0.3809 | 0.3298 |
| Treatment arm 2 | 0.0087 | 0.3828 | 0.3277 |
| Rejected arms per stage [3] | |||
| Treatment arm 1 | 0.0046 | 0.3012 | 0.3062 |
| Treatment arm 2 | 0.0140 | 0.4617 | 0.3255 |
| Success per stage [1] | 0 | 0.0020 | 0.0132 |
| Success per stage [2] | 0.0006 | 0.3041 | 0.4774 |
| Success per stage [3] | 0.0023 | 0.2982 | 0.4837 |
| Stage-wise number of subjects [1] | |||
| Treatment arm 1 | 157.0 | 114.5 | 90.7 |
| Treatment arm 2 | 157.0 | 112.9 | 91.5 |
| Control arm | 157.0 | 157.0 | 157.0 |
| Stage-wise number of subjects [2] | |||
| Treatment arm 1 | 157.0 | 147.0 | 131.1 |
| Treatment arm 2 | 157.0 | 146.9 | 132.0 |
| Control arm | 157.0 | 157.0 | 157.0 |
| Stage-wise number of subjects [3] | |||
| Treatment arm 1 | 157.0 | 139.6 | 113.2 |
| Treatment arm 2 | 157.0 | 151.8 | 145.5 |
| Control arm | 157.0 | 157.0 | 157.0 |
| Expected number of subjects under H1 [1] | 660.9 | ||
| Expected number of subjects under H1 [2] | 1146.4 | ||
| Expected number of subjects under H1 [3] | 1144.2 | ||
| Overall exit probability [1] | 0.5968 | 0.3004 | |
| Overall exit probability [2] | 0.0517 | 0.3584 | |
| Overall exit probability [3] | 0.0547 | 0.3455 | |
| Selected arms [1] | |||
| Treatment arm 1 | 1.0000 | 0.2940 | 0.0594 |
| Treatment arm 2 | 1.0000 | 0.2900 | 0.0599 |
| Selected arms [2] | |||
| Treatment arm 1 | 1.0000 | 0.8876 | 0.4926 |
| Treatment arm 2 | 1.0000 | 0.8874 | 0.4959 |
| Selected arms [3] | |||
| Treatment arm 1 | 1.0000 | 0.8405 | 0.4323 |
| Treatment arm 2 | 1.0000 | 0.9142 | 0.5557 |
| Number of active arms [1] | 2.000 | 1.448 | 1.161 |
| Number of active arms [2] | 2.000 | 1.872 | 1.676 |
| Number of active arms [3] | 2.000 | 1.856 | 1.647 |
| Conditional power (achieved) [1] | 0.0627 | 0.3925 | |
| Conditional power (achieved) [2] | 0.4348 | 0.7372 | |
| Conditional power (achieved) [3] | 0.4248 | 0.7506 | |
| Exit probability for futility [1] | 0.5968 | 0.2984 | |
| Exit probability for futility [2] | 0.0511 | 0.0543 | |
| Exit probability for futility [3] | 0.0524 | 0.0473 |
Legend:
- (i): treatment arm i
- [j]: effect matrix row j (situation to consider)
Depending on the stage of the trial, the switch() statement in the treatment arm selection function (selection()) chooses the correct futility boundaries from a predefined futility bound matrix and compares the effectVector to them. The return value of the selection function is a logical vector with one entry per active treatment arm stating whether or not the respective arm is to be selected.
Again, assuming \(H_0\) to be true (scenario [1]), the probability of falsely rejecting it is \(0.0181\), thus this simulation again indicates type I error rate control at \(\alpha=0.025\). The power assuming a \(50\%\) reduction in both active arms is \(0.8304\) (Reject at least one [2]). Since now, stopping of treatment arms is determined by crossing the futility bounds or not, the probability of a futility stop in the first stage increases to \(0.5968\) in the first and to \(0.2984\) in the second stage under \(H_0\) as compared to the first simulation. Having a different treatment selection scheme also result in lower expected number of subjects and, as opposed to the first simulation, the number of arms per scenario lies \(<2\) starting from stage \(2\) since this treatment selection allows for early discontinuation of study arms whereas in the first simulation, arms are to be carried forward regardless of potential futility.
In both simulations (using the rBest selection scheme and the user-defined selection function), with initially calculated \(157\) subjects per arm per stage and a relative event rate reduction of \(50\%\) as alternative, one can see that the study is overpowered (since Reject at least one [j]>0.8 in scenario [2]), which is due to the considered alternative having approximately the same effect in the two active treatment arms. For the second simulation using the selection function, additional simulations searching for the potentially optimal sample size (considered as the smallest sample size needed to achieve \(80\%\) power) indicate that one could save some subjects and still achieve the desired power. The following plot shows the minimum sample size that one could choose in order to achieve the power requirements of \(80\%\). The following plot displays simulated power values against sample sizes ranging from \(140\) to \(155\) per arm per stage, where the red horizontal line represents the target power of \(0.8\). The simulations indicate that the optimal sample size is around \(146\) subjects per arm and stage, however, this number may slightly deviate from an analytically optimal solution due to the nature of the simulation process:
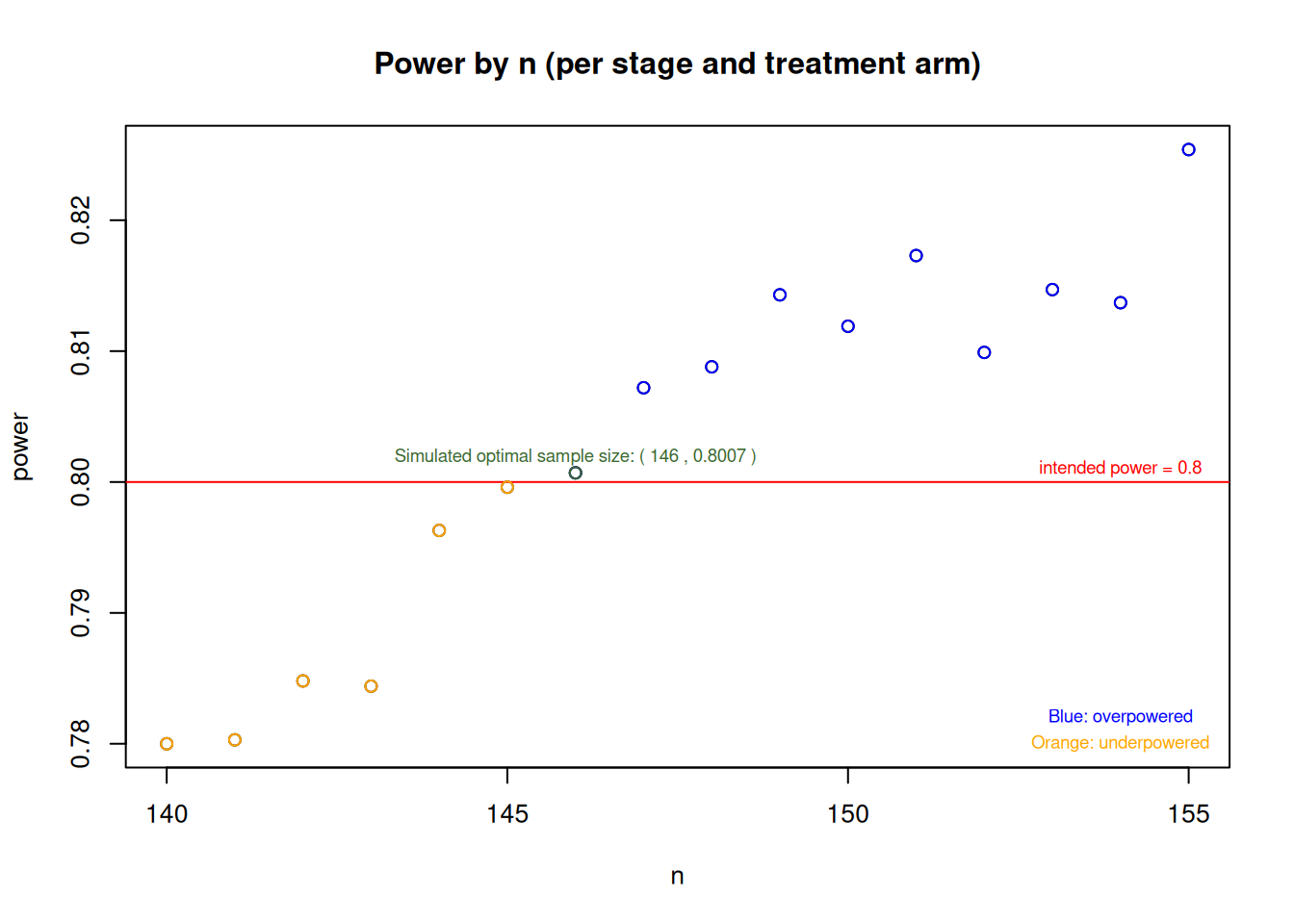
Furthermore, it should be noted here that despite having explicitly assumed \(\pi_2=0.1\) under the null hypothesis, testing the hypothesis \(H_0: \frac{\pi_1}{\pi_2}\geq1\) means that whenever \(\pi_1\geq\pi_2\) with \(\pi_2, \, \pi_1 \in (0,1]\), one has a scenario where the null hypothesis is true. Consequently, one should properly check if the type I error rate is controlled not only assuming \(\pi_2=0.1\), but also considering the other scenarios when \(\pi_2 \neq 0.1\). For the second simulation, considering only the cases where rate equality holds, the following plot obtained by simulation indicates that, given the study configurations here, the type I error rate is controlled under various control event rate assumptions:
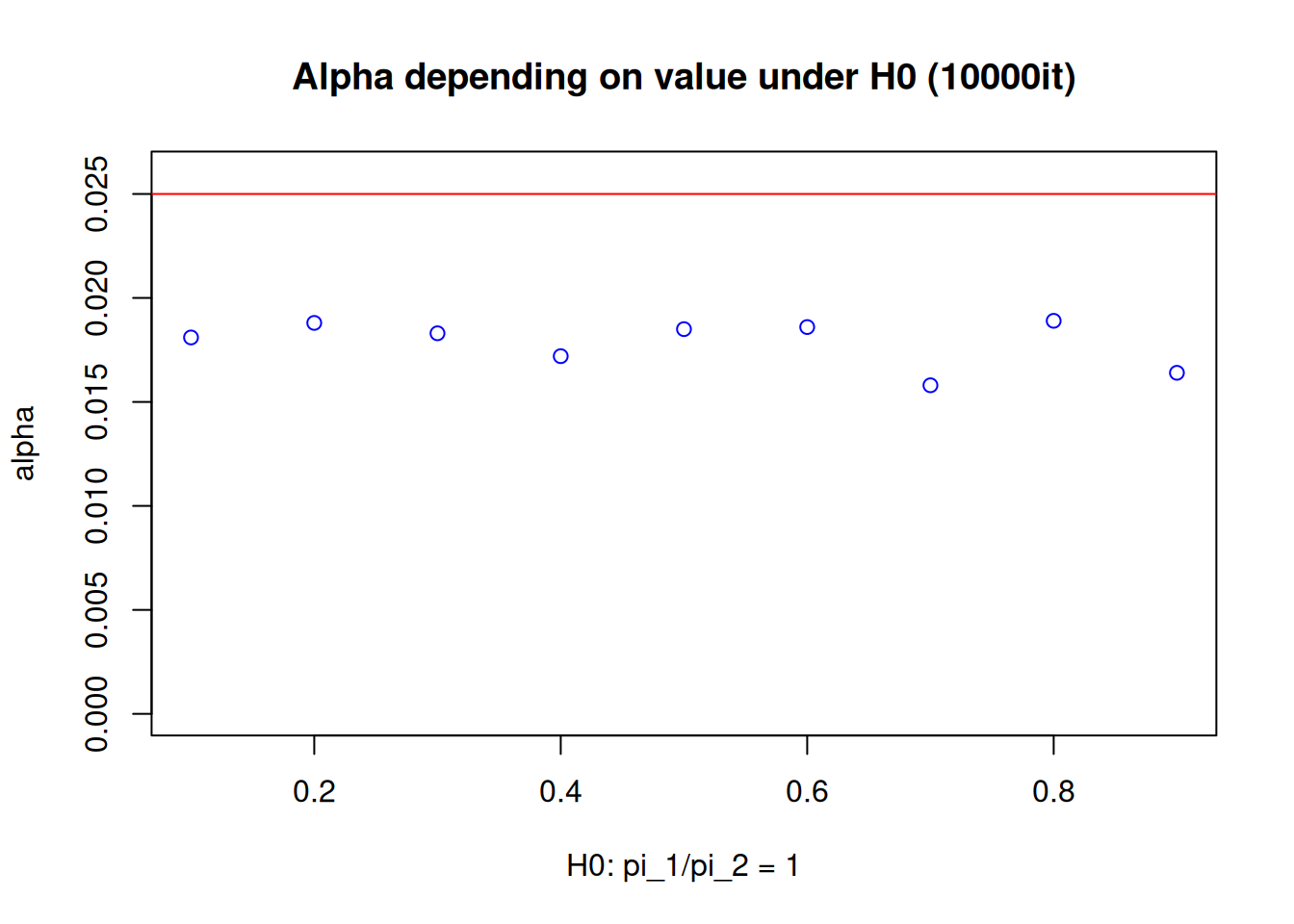
Illustrative analysis with generic Data
In the next chapters, different hypothetical binary endpoint datasets are generated and analyzed using the getAnalysisResults() command. As previously mentioned, rpact itself does not support landmark analysis using the Greenwoods standard error (SE) formula. Thus, the first two analyses are based on the empirical event rates only. Afterwards, the packages gestate and survival are then used to show how one could merge the packages to perform the intended analysis using boundaries obtained by rpact in combination with test statistics obtained by survival probabilities and standard errors estimated using gestate and survival.
First theoretical path
In the first-stage analysis, one first has to manually enter a dataset for data observed in the trial:
genData_1 <- getDataset(
events1 = 4,
events2 = 8,
events3 = 16,
sampleSizes1 = 153,
sampleSizes2 = 157,
sampleSizes3 = 156
)
genData_1 |> summary()Dataset of multi-arm rates
The dataset contains the sample sizes and events of two treatment groups and one control group.
| Stage | 1 | 1 | 1 |
|---|---|---|---|
| Group | 1 | 2 | 3 |
| Sample size | 153 | 157 | 156 |
| Number of events | 4 | 8 | 16 |
This dataset is a generic realization of data from the first stage in a design with binary endpoint under the input assumption of having a \(50\%\) rate reduction in the treatment groups given \(\pi_2=0.1\). The highest index (\(3\)) corresponds with the events and sample size of the control group. The indices \(1\) and \(2\) represent the active treatment groups. The data here is chosen such that one can see lower event rates in the active treatment groups. Note the slight imbalances in sample sizes, which may occur due to dropouts or recruitment issues.
The analysis of the first stage using Simes as intersection test is performed as follows:
results_1 <- getAnalysisResults(
design = d_IN,
dataInput = genData_1,
directionUpper = FALSE,
intersectionTest = "Simes"
)results_1 |> summary()Multi-arm analysis results for a binary endpoint (2 active arms vs. control)
Sequential analysis with 3 looks (inverse normal combination test design), one-sided overall significance level 2.5%. The results were calculated using a multi-arm test for rates, Simes intersection test, normal approximation test. H0: pi(i) - pi(control) = 0 against H1: pi(i) - pi(control) < 0.
| Stage | 1 | 2 | 3 |
|---|---|---|---|
| Fixed weight | 0.577 | 0.577 | 0.577 |
| Cumulative alpha spent | 0.0001 | 0.0060 | 0.0250 |
| Stage levels (one-sided) | 0.0001 | 0.0060 | 0.0231 |
| Efficacy boundary (z-value scale) | 3.710 | 2.511 | 1.993 |
| Futility boundary (z-value scale) | 0.149 | 0.414 | |
| Cumulative effect size (1) | -0.076 | ||
| Cumulative effect size (2) | -0.052 | ||
| Cumulative treatment rate (1) | 0.026 | ||
| Cumulative treatment rate (2) | 0.051 | ||
| Cumulative control rate | 0.103 | ||
| Stage-wise test statistic (1) | -2.730 | ||
| Stage-wise test statistic (2) | -1.716 | ||
| Stage-wise p-value (1) | 0.0032 | ||
| Stage-wise p-value (2) | 0.0431 | ||
| Adjusted stage-wise p-value (1, 2) | 0.0063 | ||
| Adjusted stage-wise p-value (1) | 0.0032 | ||
| Adjusted stage-wise p-value (2) | 0.0431 | ||
| Overall adjusted test statistic (1, 2) | 2.493 | ||
| Overall adjusted test statistic (1) | 2.730 | ||
| Overall adjusted test statistic (2) | 1.716 | ||
| Test action: reject (1) | FALSE | ||
| Test action: reject (2) | FALSE | ||
| Conditional rejection probability (1) | 0.2907 | ||
| Conditional rejection probability (2) | 0.1204 | ||
| 95% repeated confidence interval (1) | [-0.212; 0.043] | ||
| 95% repeated confidence interval (2) | [-0.191; 0.079] | ||
| Repeated p-value (1) | 0.1150 | ||
| Repeated p-value (2) | 0.2429 |
Legend:
- (i): results of treatment arm i vs. control arm
- (i, j, …): comparison of treatment arms ‘i, j, …’ vs. control arm
Although lower event rates in the active arms were observed in the first stage, neither of the \(2\) hypotheses can be rejected at this point. This can be seen by e.g., comparing the overall adjusted test statistic for the global intersection test (which tests the null hypothesis that there is no effect in either active treatment arm) to the efficacy boundary. Since \(2.493 < 3.710\), we cannot reject after the first stage. Neither of the futility boundaries is crossed at stage \(1\), which means that both treatment arms are to be carried forward to the second stage. For obtaining non-significance in stage \(1\), one could also compare repeated p-values to full \(\alpha\).
Proceeding to the second stage, a generic dataset might look as follows, with the second vector entries represent the second-stage data:
# assuming there was no futility or efficacy stop study proceeds to randomize subjects
genData_2 <- getDataset(
events1 = c(4, 7),
events2 = c(8, 7),
events3 = c(16, 15),
sampleSizes1 = c(153, 155),
sampleSizes2 = c(157, 155),
sampleSizes3 = c(156, 155)
)
genData_2 |> summary()Dataset of multi-arm rates
The dataset contains the sample sizes and events of two treatment groups and one control group. The total number of looks is two; stage-wise and cumulative data are included.
| Stage | 1 | 1 | 1 | 2 | 2 | 2 |
|---|---|---|---|---|---|---|
| Group | 1 | 2 | 3 | 1 | 2 | 3 |
| Stage-wise sample size | 153 | 157 | 156 | 155 | 155 | 155 |
| Cumulative sample size | 153 | 157 | 156 | 308 | 312 | 311 |
| Stage-wise number of events | 4 | 8 | 16 | 7 | 7 | 15 |
| Cumulative number of events | 4 | 8 | 16 | 11 | 15 | 31 |
Here, again, the observed event rates are lower in the treatment groups in comparison to the control group.
results_2 <- getAnalysisResults(
design = d_IN,
dataInput = genData_2,
directionUpper = FALSE,
intersectionTest = "Simes"
)results_2 |> summary()Multi-arm analysis results for a binary endpoint (2 active arms vs. control)
Sequential analysis with 3 looks (inverse normal combination test design), one-sided overall significance level 2.5%. The results were calculated using a multi-arm test for rates, Simes intersection test, normal approximation test. H0: pi(i) - pi(control) = 0 against H1: pi(i) - pi(control) < 0.
| Stage | 1 | 2 | 3 |
|---|---|---|---|
| Fixed weight | 0.577 | 0.577 | 0.577 |
| Cumulative alpha spent | 0.0001 | 0.0060 | 0.0250 |
| Stage levels (one-sided) | 0.0001 | 0.0060 | 0.0231 |
| Efficacy boundary (z-value scale) | 3.710 | 2.511 | 1.993 |
| Futility boundary (z-value scale) | 0.149 | 0.414 | |
| Cumulative effect size (1) | -0.076 | -0.064 | |
| Cumulative effect size (2) | -0.052 | -0.052 | |
| Cumulative treatment rate (1) | 0.026 | 0.036 | |
| Cumulative treatment rate (2) | 0.051 | 0.048 | |
| Cumulative control rate | 0.103 | 0.100 | |
| Stage-wise test statistic (1) | -2.730 | -1.770 | |
| Stage-wise test statistic (2) | -1.716 | -1.770 | |
| Stage-wise p-value (1) | 0.0032 | 0.0384 | |
| Stage-wise p-value (2) | 0.0431 | 0.0384 | |
| Adjusted stage-wise p-value (1, 2) | 0.0063 | 0.0384 | |
| Adjusted stage-wise p-value (1) | 0.0032 | 0.0384 | |
| Adjusted stage-wise p-value (2) | 0.0431 | 0.0384 | |
| Overall adjusted test statistic (1, 2) | 2.493 | 3.014 | |
| Overall adjusted test statistic (1) | 2.730 | 3.182 | |
| Overall adjusted test statistic (2) | 1.716 | 2.464 | |
| Test action: reject (1) | FALSE | TRUE | |
| Test action: reject (2) | FALSE | FALSE | |
| Conditional rejection probability (1) | 0.2907 | 0.7911 | |
| Conditional rejection probability (2) | 0.1204 | 0.5133 | |
| 95% repeated confidence interval (1) | [-0.212; 0.043] | [-0.130; -0.005] | |
| 95% repeated confidence interval (2) | [-0.191; 0.079] | [-0.119; 0.011] | |
| Repeated p-value (1) | 0.1150 | 0.0086 | |
| Repeated p-value (2) | 0.2429 | 0.0274 |
Legend:
- (i): results of treatment arm i vs. control arm
- (i, j, …): comparison of treatment arms ‘i, j, …’ vs. control arm
Performing the same comparisons as in stage \(1\), one can see: The global null is to be rejected (\(3.014 > 2.511\)). Subsequently, the hypothesis for the first active treatment is rejected, since \(3.182 > 2.511\) indicates that the first active arm performs better than control. The same result can be obtained by recognizing that the repeated p-value (1) of \(0.0086\) falls below \(0.025\). The consequence is that this arm can be discontinued early due to efficacy. However, since no significant result was observed for the second treatment arm (\(2.464 < 2.511\)) and an early end of the trial due to efficacy is only allowed in case all active treatment arms are significant, the study will continue to the third and final stage with the second treatment arm only. Again, no futility stop occurs. It should be noted here that the adjusted stage-wise p-values cannot directly be used for testing, but the values are based on the second-stage data only. Since an inverse normal combination test is performed with equal weights for the stages (i.e., weights of \(\sqrt{\frac{1}{3}}\) here), the first- and second-stage p-values can be used to calculate the overall adjusted test statistics, namely \(\frac{\sqrt{\frac{1}{3}} \cdot \Phi^{-1}(1-0.0063)+\sqrt{\frac{1}{3}} \cdot \Phi^{-1}(1-0.0384)}{\sqrt{\frac{2}{3}}} \approx 3.014\).
The data collected at the final stage may look like the following, where the NA entries in the first active arm indicate that no additional patients were recruited here:
genData_3 <- getDataset(
events1 = c(4, 7, NA),
events2 = c(8, 7, 6),
events3 = c(16, 15, 16),
sampleSizes1 = c(153, 155, NA),
sampleSizes2 = c(157, 155, 156),
sampleSizes3 = c(156, 155, 160)
)
genData_3 |> summary()Dataset of multi-arm rates
The dataset contains the sample sizes and events of two treatment groups and one control group. The total number of looks is three; stage-wise and cumulative data are included.
| Stage | 1 | 1 | 1 | 2 | 2 | 2 | 3 | 3 | 3 |
|---|---|---|---|---|---|---|---|---|---|
| Group | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 |
| Stage-wise sample size | 153 | 157 | 156 | 155 | 155 | 155 | NA | 156 | 160 |
| Cumulative sample size | 153 | 157 | 156 | 308 | 312 | 311 | NA | 468 | 471 |
| Stage-wise number of events | 4 | 8 | 16 | 7 | 7 | 15 | NA | 6 | 16 |
| Cumulative number of events | 4 | 8 | 16 | 11 | 15 | 31 | NA | 21 | 47 |
The results from the final analysis are:
results_3 <- getAnalysisResults(
design = d_IN,
dataInput = genData_3,
directionUpper = FALSE,
intersectionTest = "Simes"
)results_3 |> summary()Multi-arm analysis results for a binary endpoint (2 active arms vs. control)
Sequential analysis with 3 looks (inverse normal combination test design), one-sided overall significance level 2.5%. The results were calculated using a multi-arm test for rates, Simes intersection test, normal approximation test. H0: pi(i) - pi(control) = 0 against H1: pi(i) - pi(control) < 0.
| Stage | 1 | 2 | 3 |
|---|---|---|---|
| Fixed weight | 0.577 | 0.577 | 0.577 |
| Cumulative alpha spent | 0.0001 | 0.0060 | 0.0250 |
| Stage levels (one-sided) | 0.0001 | 0.0060 | 0.0231 |
| Efficacy boundary (z-value scale) | 3.710 | 2.511 | 1.993 |
| Futility boundary (z-value scale) | 0.149 | 0.414 | |
| Cumulative effect size (1) | -0.076 | -0.064 | |
| Cumulative effect size (2) | -0.052 | -0.052 | -0.055 |
| Cumulative treatment rate (1) | 0.026 | 0.036 | |
| Cumulative treatment rate (2) | 0.051 | 0.048 | 0.045 |
| Cumulative control rate | 0.103 | 0.100 | 0.100 |
| Stage-wise test statistic (1) | -2.730 | -1.770 | |
| Stage-wise test statistic (2) | -1.716 | -1.770 | -2.149 |
| Stage-wise p-value (1) | 0.0032 | 0.0384 | |
| Stage-wise p-value (2) | 0.0431 | 0.0384 | 0.0158 |
| Adjusted stage-wise p-value (1, 2) | 0.0063 | 0.0384 | 0.0158 |
| Adjusted stage-wise p-value (1) | 0.0032 | 0.0384 | |
| Adjusted stage-wise p-value (2) | 0.0431 | 0.0384 | 0.0158 |
| Overall adjusted test statistic (1, 2) | 2.493 | 3.014 | 3.702 |
| Overall adjusted test statistic (1) | 2.730 | 3.182 | |
| Overall adjusted test statistic (2) | 1.716 | 2.464 | 3.253 |
| Test action: reject (1) | FALSE | TRUE | TRUE |
| Test action: reject (2) | FALSE | FALSE | TRUE |
| Conditional rejection probability (1) | 0.2907 | 0.7911 | |
| Conditional rejection probability (2) | 0.1204 | 0.5133 | |
| 95% repeated confidence interval (1) | [-0.212; 0.043] | [-0.130; -0.005] | |
| 95% repeated confidence interval (2) | [-0.191; 0.079] | [-0.119; 0.011] | [-0.099; -0.013] |
| Repeated p-value (1) | 0.1150 | 0.0086 | |
| Repeated p-value (2) | 0.2429 | 0.0274 | 0.0006 |
Legend:
- (i): results of treatment arm i vs. control arm
- (i, j, …): comparison of treatment arms ‘i, j, …’ vs. control arm
After the third stage, we can see that the null hypothesis of the second active treatment arm can now also be rejected. This can be seen by considering the overall adjusted test statistics for the intersection and single hypothesis tests, the repeated p-values or by directly referring to Test action: reject (2). Since now, both the first and second active treatment arm have been observed to be superior to control, the study concludes with success.
The results can be visualized by plotting the repeated confidence intervals, which become narrower with each stage due to the increasing cumulative sample sizes and consistent effect trend observed across stages:
results_3 |> plot(type = 2)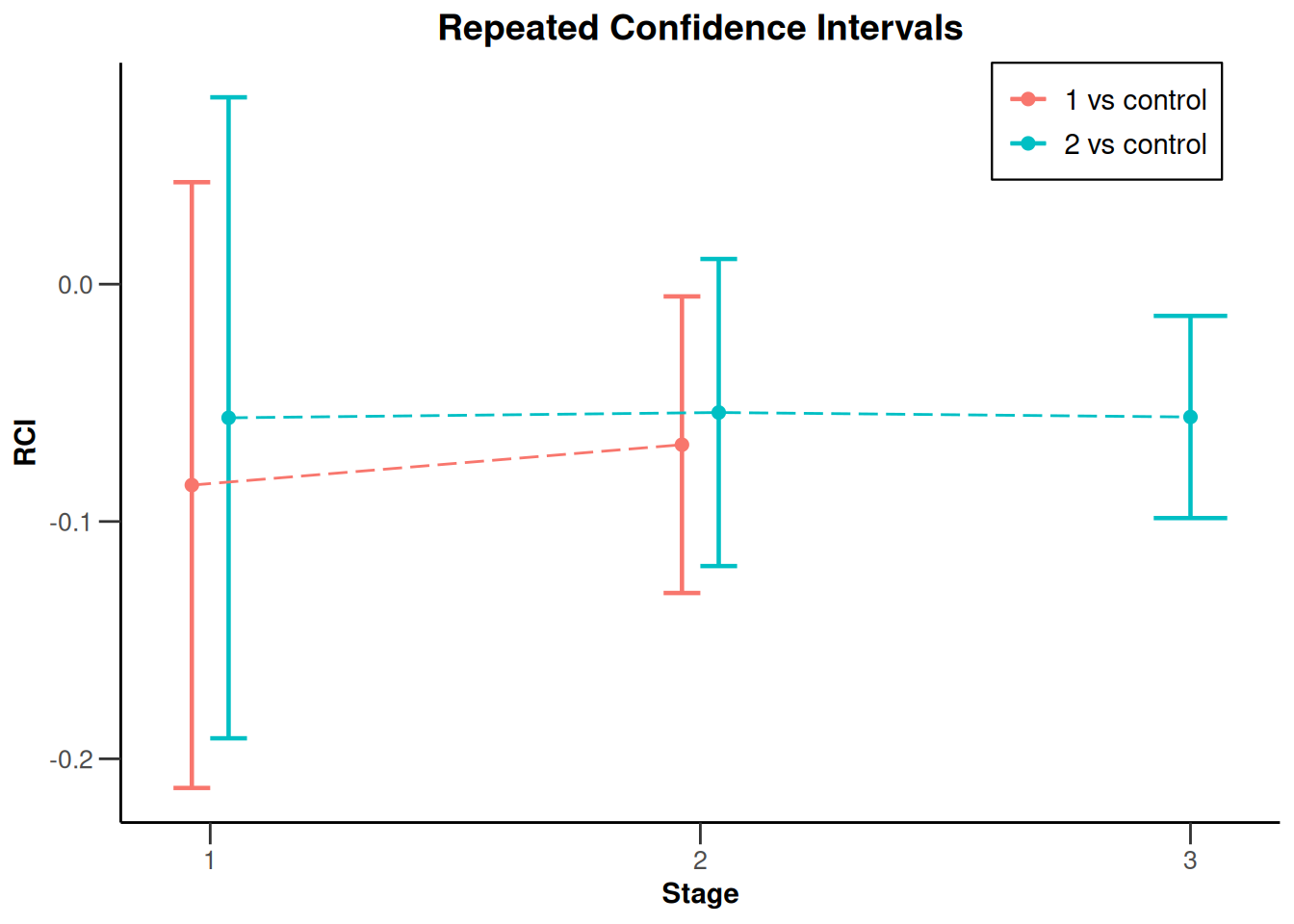
Second theoretical path
For the second analysis example, assume that we observe the same first-stage dataset (and thus, the same analysis results) as above, but the second-stage dataset is now:
genData_4 <- getDataset(
events1 = c(4, 9),
events2 = c(8, 23),
events3 = c(16, 15),
sampleSizes1 = c(153, 155),
sampleSizes2 = c(157, 155),
sampleSizes3 = c(156, 155)
)Note the large number of events in the second active treatment arm in the second stage.
Using this dataset for analysis yields the following results:
results_4 <- getAnalysisResults(
design = d_IN,
dataInput = genData_4,
directionUpper = FALSE,
intersectionTest = "Simes"
)results_4 |> summary()Multi-arm analysis results for a binary endpoint (2 active arms vs. control)
Sequential analysis with 3 looks (inverse normal combination test design), one-sided overall significance level 2.5%. The results were calculated using a multi-arm test for rates, Simes intersection test, normal approximation test. H0: pi(i) - pi(control) = 0 against H1: pi(i) - pi(control) < 0.
| Stage | 1 | 2 | 3 |
|---|---|---|---|
| Fixed weight | 0.577 | 0.577 | 0.577 |
| Cumulative alpha spent | 0.0001 | 0.0060 | 0.0250 |
| Stage levels (one-sided) | 0.0001 | 0.0060 | 0.0231 |
| Efficacy boundary (z-value scale) | 3.710 | 2.511 | 1.993 |
| Futility boundary (z-value scale) | 0.149 | 0.414 | |
| Cumulative effect size (1) | -0.076 | -0.057 | |
| Cumulative effect size (2) | -0.052 | 0.000 | |
| Cumulative treatment rate (1) | 0.026 | 0.042 | |
| Cumulative treatment rate (2) | 0.051 | 0.099 | |
| Cumulative control rate | 0.103 | 0.100 | |
| Stage-wise test statistic (1) | -2.730 | -1.275 | |
| Stage-wise test statistic (2) | -1.716 | 1.385 | |
| Stage-wise p-value (1) | 0.0032 | 0.1011 | |
| Stage-wise p-value (2) | 0.0431 | 0.9170 | |
| Adjusted stage-wise p-value (1, 2) | 0.0063 | 0.2023 | |
| Adjusted stage-wise p-value (1) | 0.0032 | 0.1011 | |
| Adjusted stage-wise p-value (2) | 0.0431 | 0.9170 | |
| Overall adjusted test statistic (1, 2) | 2.493 | 2.352 | |
| Overall adjusted test statistic (1) | 2.730 | 2.832 | |
| Overall adjusted test statistic (2) | 1.716 | 0.234 | |
| Test action: reject (1) | FALSE | FALSE | |
| Test action: reject (2) | FALSE | FALSE | |
| Conditional rejection probability (1) | 0.2907 | 0.4500 | |
| Conditional rejection probability (2) | 0.1204 | 0.0009 | |
| 95% repeated confidence interval (1) | [-0.212; 0.043] | [-0.125; 0.003] | |
| 95% repeated confidence interval (2) | [-0.191; 0.079] | [-0.080; 0.075] | |
| Repeated p-value (1) | 0.1150 | 0.0340 | |
| Repeated p-value (2) | 0.2429 | 0.2429 |
Legend:
- (i): results of treatment arm i vs. control arm
- (i, j, …): comparison of treatment arms ‘i, j, …’ vs. control arm
As we can see, the global null hypothesis cannot be rejected at the second stage, since \(2.352 < 2.511\). However, the second active treatment arm is stopped for futility, since, after not rejecting the global null, its corresponding overall adjusted test statistic of \(0.234\) is smaller than the second-stage futility bound of \(0.414.\) Therefore, this study course leads to a continuation to the third stage with active treatment arm \(1\) only.
As the final-stage dataset, we may then observe:
genData_5 <- getDataset(
events1 = c(4, 9, 7),
events2 = c(8, 23, NA),
events3 = c(16, 15, 16),
sampleSizes1 = c(153, 155, 165),
sampleSizes2 = c(157, 155, NA),
sampleSizes3 = c(156, 155, 160)
)
genData_5 |> summary()Dataset of multi-arm rates
The dataset contains the sample sizes and events of two treatment groups and one control group. The total number of looks is three; stage-wise and cumulative data are included.
| Stage | 1 | 1 | 1 | 2 | 2 | 2 | 3 | 3 | 3 |
|---|---|---|---|---|---|---|---|---|---|
| Group | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 |
| Stage-wise sample size | 153 | 157 | 156 | 155 | 155 | 155 | 165 | NA | 160 |
| Cumulative sample size | 153 | 157 | 156 | 308 | 312 | 311 | 473 | NA | 471 |
| Stage-wise number of events | 4 | 8 | 16 | 9 | 23 | 15 | 7 | NA | 16 |
| Cumulative number of events | 4 | 8 | 16 | 13 | 31 | 31 | 20 | NA | 47 |
Note that recruitment for treatment arm \(2\) has been terminated after stage \(2\) and NA is entered for the respective vectors.
The final stage analysis leads to the results:
results_5 <- getAnalysisResults(
design = d_IN,
dataInput = genData_5,
directionUpper = FALSE,
intersectionTest = "Simes"
)results_5 |> summary()Multi-arm analysis results for a binary endpoint (2 active arms vs. control)
Sequential analysis with 3 looks (inverse normal combination test design), one-sided overall significance level 2.5%. The results were calculated using a multi-arm test for rates, Simes intersection test, normal approximation test. H0: pi(i) - pi(control) = 0 against H1: pi(i) - pi(control) < 0.
| Stage | 1 | 2 | 3 |
|---|---|---|---|
| Fixed weight | 0.577 | 0.577 | 0.577 |
| Cumulative alpha spent | 0.0001 | 0.0060 | 0.0250 |
| Stage levels (one-sided) | 0.0001 | 0.0060 | 0.0231 |
| Efficacy boundary (z-value scale) | 3.710 | 2.511 | 1.993 |
| Futility boundary (z-value scale) | 0.149 | 0.414 | |
| Cumulative effect size (1) | -0.076 | -0.057 | -0.058 |
| Cumulative effect size (2) | -0.052 | 0.000 | |
| Cumulative treatment rate (1) | 0.026 | 0.042 | 0.042 |
| Cumulative treatment rate (2) | 0.051 | 0.099 | |
| Cumulative control rate | 0.103 | 0.100 | 0.100 |
| Stage-wise test statistic (1) | -2.730 | -1.275 | -2.024 |
| Stage-wise test statistic (2) | -1.716 | 1.385 | |
| Stage-wise p-value (1) | 0.0032 | 0.1011 | 0.0215 |
| Stage-wise p-value (2) | 0.0431 | 0.9170 | |
| Adjusted stage-wise p-value (1, 2) | 0.0063 | 0.2023 | 0.0215 |
| Adjusted stage-wise p-value (1) | 0.0032 | 0.1011 | 0.0215 |
| Adjusted stage-wise p-value (2) | 0.0431 | 0.9170 | |
| Overall adjusted test statistic (1, 2) | 2.493 | 2.352 | 3.089 |
| Overall adjusted test statistic (1) | 2.730 | 2.832 | 3.481 |
| Overall adjusted test statistic (2) | 1.716 | 0.234 | |
| Test action: reject (1) | FALSE | FALSE | TRUE |
| Test action: reject (2) | FALSE | FALSE | FALSE |
| Conditional rejection probability (1) | 0.2907 | 0.4500 | |
| Conditional rejection probability (2) | 0.1204 | 0.0009 | |
| 95% repeated confidence interval (1) | [-0.212; 0.043] | [-0.125; 0.003] | [-0.102; -0.017] |
| 95% repeated confidence interval (2) | [-0.191; 0.079] | [-0.080; 0.075] | |
| Repeated p-value (1) | 0.1150 | 0.0340 | 0.0010 |
| Repeated p-value (2) | 0.2429 | 0.2429 |
Legend:
- (i): results of treatment arm i vs. control arm
- (i, j, …): comparison of treatment arms ‘i, j, …’ vs. control arm
Since per definition, success of one treatment arm suffices (because there is only one treatment arm left) to declare study success, that is the conclusion one comes to due to significance of treatment arm \(1\). Stopping treatment arm \(2\) earlier therefore did not influence the positive result.
Landmark analysis with Greenwood’s formula
A landmark analysis is an analysis where, given a predefined and fixed point in time, survival probabilities at that specific time point are to be compared between different groups, such as active treatment and control group.
As previously mentioned, rpact itself does not support the procedure of comparing survival probabilities (more precisely, their estimates) at a specific point in time with the standard error being calculated by Greenwoods formula, but is based on simple rate comparisons only. However, the R package gestate supports various survival analysis methods, including landmark analysis. Firstly, one needs to load the package:
library(gestate)Then, since the functionality of gestate is based on curve subjects and the analysis approach is based on survival data, the distribution of the data of the treatment and control groups need to be specified. Assume that the data in the control group follows an exponential distribution Exp(\(\lambda_c\)) with \(\lambda_c=0.1\) and the treatment groups follow Exp(\(\lambda_{t1}\))- and Exp(\(\lambda_{t2}\))-distributions with \(\lambda_{t1}=\lambda_{t2}=0.1\cdot0.5=0.05\) respectively (hazard ratio of \(0.5\), heuristically representing the assumed treatment effect of a \(50\%\) rate-reduction), while other options would be to assume Weibull or piecewise Exponential distribution. This is initialized using:
effect <- 0.5
# initializing assumed distributions of different treatment arms as well as control
dist_c <- gestate::Exponential(lambda = 0.1)
dist_t1 <- gestate::Exponential(lambda = 0.1 * (1 - effect))
dist_t2 <- gestate::Exponential(lambda = 0.1 * (1 - effect))Then, another curve that needs to be initialized is the recruitment curve. gestate covers the potential assumptions of, e.g., having instantaneous, linear or piecewise linear recruitment. In many applications, assuming linear recruitment (i.e., a constant recruitment rate) is generally suitable and will be used for illustration here. We will assume the same sample sizes as calculated in chapter \(2\) and, since measuring study duration in months is common, one may assume a total recruitment length of \(18\) months = \(1.5\) years. The data will later on be used to estimate survival probabilities and standard errors, subsequently to be used as input for the rpact continuous endpoint module. Since the mean, standard deviation and sample sizes in the dataset input all need to be stage-wise because of the use of inverse normal combination test, data simulation will also be done separately per stage per arm, resulting in a maximum of 9 simulated datasets in this application. Furthermore, since the information levels are equally spread along the three stages, each stage-wise dataset will be based on a recruitment of \(157\) subjects and a recruitment duration of \(6\) months. Assuming an individual observation time of \(1\) month, the total duration of each stage is \(7\) months:
# equally sized arms and stages
n <- 157
# maximum duration of each stage
maxtime <- 7
# initialize recruitment for control group
recruitment_c <- gestate::LinearR(Nactive = 0, Ncontrol = n, rlength = 6)
# initialize recruitment for treatment groups, here: equal
recruitment_t1 <- gestate::LinearR(Nactive = n, Ncontrol = 0, rlength = 6)
recruitment_t2 <- gestate::LinearR(Nactive = n, Ncontrol = 0, rlength = 6)As already mentioned in chapter \(1\) (initialization of design), the interim analyses are to be performed at \(I_1 = \frac{1}{3}\) and \(I_2 = \frac{2}{3}\) with \(I\) denoting the information fraction and the index denoting the stage. Since information is directly correlated with sample size here, one can conclude that interim analyses should be performed after \(6\) months and after \(12\) months of recruitment. Censoring could be specified via the variables active_dcurve, control_dcurve, however, in trials in which no or only little censoring might be a reasonable assumption, the default values for these variables can be used (=Blank()). The only remaining type of censoring is censoring due to end of observation, i.e., subjects having had no event before study ends are considered censored.
Having made assumptions on data distribution, recruitment and censoring, simulating patient level survival data works as follows. It should be noted here that the following simulations and data manipulations are demonstrated for the first stage data, however, the process is similar for the second and third stage.
# simulating data for all arms and stages independently (needed for dataset input)
# first stage, all arms, last index represents the stage
example_data_long_t1_1 <- gestate::simulate_trials(
active_ecurve = dist_t1,
control_ecurve = Blank(),
rcurve = recruitment_t1,
assess = maxtime,
iterations = 1,
seed = 1,
detailed_output = TRUE
)
example_data_long_t2_1 <- gestate::simulate_trials(
active_ecurve = dist_t2,
control_ecurve = Blank(),
rcurve = recruitment_t2,
assess = maxtime,
iterations = 1,
seed = 2,
detailed_output = TRUE
)
example_data_long_c_1 <- gestate::simulate_trials(
active_ecurve = Blank(),
control_ecurve = dist_c,
rcurve = recruitment_c,
assess = maxtime,
iterations = 1,
seed = 3,
detailed_output = TRUE
)
kable(head(example_data_long_t1_1, 10))| Time | Censored | Trt | Iter | ETime | CTime | Rec_Time | Assess | Max_F | RCTime |
|---|---|---|---|---|---|---|---|---|---|
| 4.136118 | 1 | 2 | 1 | 15.103637 | Inf | 2.8638823 | 7 | Inf | 4.136118 |
| 2.375578 | 1 | 2 | 1 | 23.632856 | Inf | 4.6244223 | 7 | Inf | 2.375578 |
| 2.914134 | 0 | 2 | 1 | 2.914134 | Inf | 0.1667227 | 7 | Inf | 6.833277 |
| 2.795905 | 0 | 2 | 1 | 2.795905 | Inf | 3.1638647 | 7 | Inf | 3.836135 |
| 1.718086 | 1 | 2 | 1 | 8.721372 | Inf | 5.2819144 | 7 | Inf | 1.718086 |
| 4.761620 | 1 | 2 | 1 | 57.899371 | Inf | 2.2383802 | 7 | Inf | 4.761620 |
| 6.712245 | 1 | 2 | 1 | 24.591241 | Inf | 0.2877548 | 7 | Inf | 6.712245 |
| 6.168230 | 1 | 2 | 1 | 10.793657 | Inf | 0.8317695 | 7 | Inf | 6.168230 |
| 5.071047 | 1 | 2 | 1 | 19.131350 | Inf | 1.9289527 | 7 | Inf | 5.071047 |
| 2.940920 | 0 | 2 | 1 | 2.940920 | Inf | 0.9289897 | 7 | Inf | 6.071010 |
The table above provides information on patient level, such as time (to event), treatment group, time of event (if observed), assessment timing and recruitment time. To achieve that every subject only has an individual observation time of \(1\) month, one can manually set the censoring indicator to \(1\) whenever no event has happened or the simulation indicates that an event has happened only after the fixed observation time.
# Check: if time to event (first column) is >=1, subject is censored
example_data_long_t1_1[which(example_data_long_t1_1[, 1] > 1), 2] <- 1
example_data_long_t2_1[which(example_data_long_t2_1[, 1] > 1), 2] <- 1
example_data_long_c_1[which(example_data_long_c_1[, 1] > 1), 2] <- 1After doing this, since the administrative censoring due to end of observation has been manually manipulated, the time of censoring in the last column can manually be corrected according to the changes:
# if censoring indicator==1, the censoring time is set to event time
# according to the previous changes
# stage 1
example_data_long_t1_1[which(example_data_long_t1_1[, 2] == 1), 9] <-
example_data_long_t1_1[which(example_data_long_t1_1[, 2] == 1), 1]
example_data_long_t2_1[which(example_data_long_t2_1[, 2] == 1), 9] <-
example_data_long_t2_1[which(example_data_long_t2_1[, 2] == 1), 1]
example_data_long_c_1[which(example_data_long_c_1[, 2] == 1), 9] <-
example_data_long_c_1[which(example_data_long_c_1[, 2] == 1), 1]Though working on subject level data (if available) is commonly recommended, one could be interested in tables containing time, number of subjects at risk, survival probabilities and respective standard errors, henceforth referred to as life tables. After setting the assessment time to the defined \(7\) months, for creating the life tables, the commands Surv() and survfit() from the R package survival are used:
# needs to be run in case package has not yet been installed
library(survival)
# setting desired assessment time
example_data_short_t1_1 <- gestate::set_assess_time(
example_data_long_t1_1,
maxtime
)
example_data_short_t2_1 <- gestate::set_assess_time(
example_data_long_t2_1,
maxtime
)
example_data_short_c_1 <- gestate::set_assess_time(
example_data_long_c_1,
maxtime
)
# Creating live tables for each group depending on the stage
# at first landmark time point
lt_t1_1 <- summary(survival::survfit(survival::Surv(
example_data_short_t1_1[, "Time"],
1 - example_data_short_t1_1[, "Censored"]
) ~ 1, error = "greenwood"))
lt_t2_1 <- summary(survival::survfit(survival::Surv(
example_data_short_t2_1[, "Time"],
1 - example_data_short_t2_1[, "Censored"]
) ~ 1, error = "greenwood"))
lt_c_1 <- summary(survival::survfit(survival::Surv(
example_data_short_c_1[, "Time"],
1 - example_data_short_c_1[, "Censored"]
) ~ 1, error = "greenwood"))
kable(head(cbind(
"Time" = lt_t1_1$time, "n.risk" = lt_t1_1$n.risk,
"n.event" = lt_t1_1$n.event, "surv.prob." = lt_t1_1$surv,
"Greenwoods SE" = lt_t1_1$std.err
), 10))| Time | n.risk | n.event | surv.prob. | Greenwoods SE |
|---|---|---|---|---|
| 0.4050083 | 157 | 1 | 0.9936306 | 0.0063491 |
| 0.7430455 | 156 | 1 | 0.9872611 | 0.0089502 |
| 0.7453705 | 155 | 1 | 0.9808917 | 0.0109263 |
| 1.0411090 | 152 | 1 | 0.9744385 | 0.0126170 |
| 1.1852241 | 143 | 1 | 0.9676242 | 0.0142506 |
| 1.1887832 | 142 | 1 | 0.9608100 | 0.0156951 |
| 1.3882469 | 136 | 1 | 0.9537452 | 0.0170959 |
| 1.7934816 | 127 | 1 | 0.9462354 | 0.0185375 |
| 1.8221775 | 125 | 1 | 0.9386655 | 0.0198748 |
| 1.9769123 | 119 | 1 | 0.9307776 | 0.0212154 |
The above table contains time, number of subjects at risk, number of events, survival probability estimation and standard error estimation, thus allowing for the creation of a Kaplan-Meier-Plot to visualize the development of survival probability in time. Note that choosing error = "greenwood" leads to the desired SE estimation. One could also discretize the time into intervals to obtain life tables with consistent time steps, if desired. Exemplary Kaplan-Meier-Plots for the different treatment groups at the different stages are displayed below:
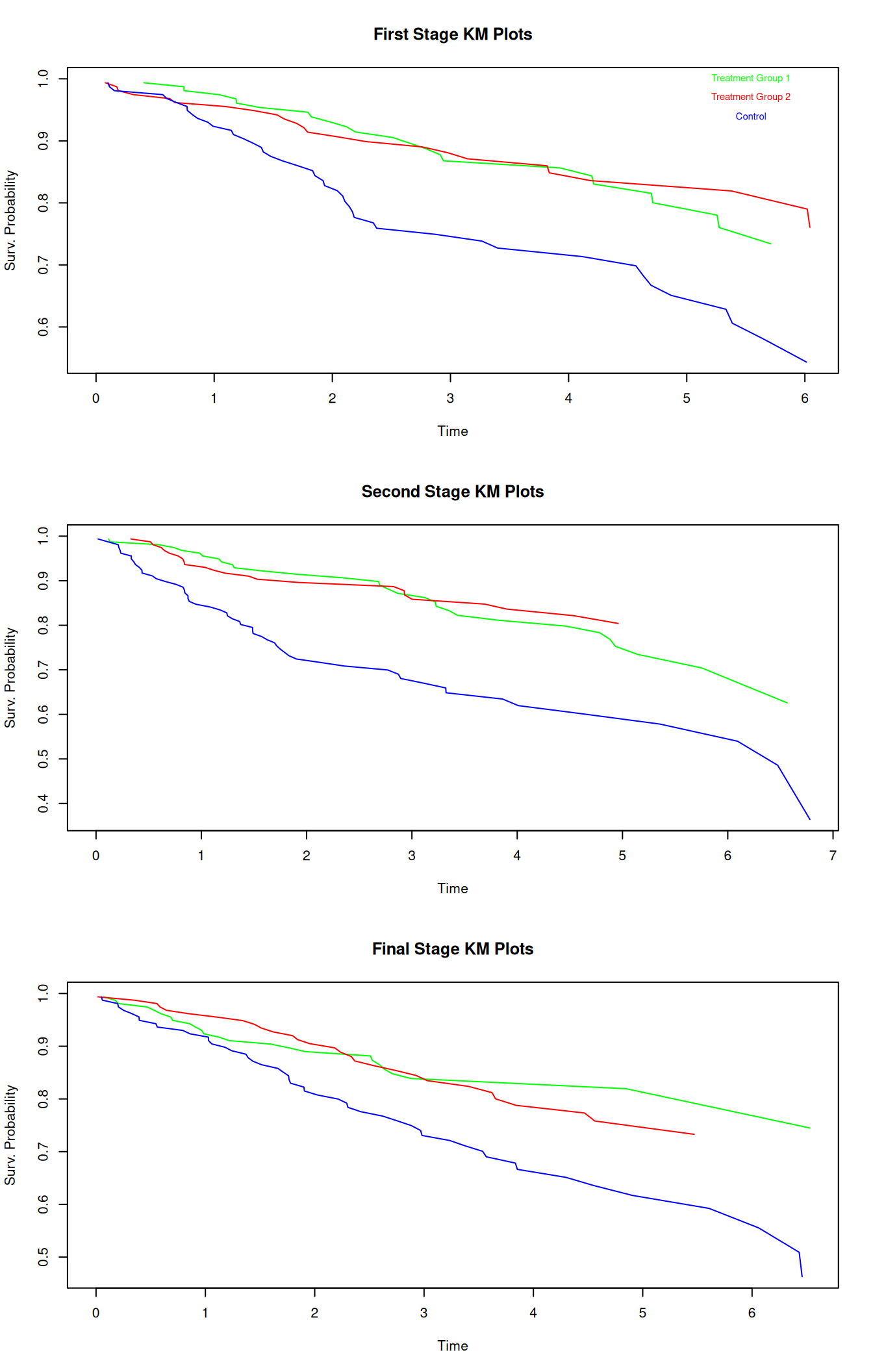
While the survival probabilities of the treatment arms develop rather similarly for the independently simulated stages, the figures demonstrate that the survival probabilities of the control group almost always fall below the survival probabilities of the treatment groups (apart from a few early exceptions), indicating that the groups may (i.e. potentially statistically significantly) differ.
As previously mentioned, the continuous module of rpact is now used to perform the landmark analysis of the gestate-simulated data. Note that this approximation is valid whenever it is reasonable to assume that the survival probabilities are normally distributed. If this assumption holds, we can initialize continuous stage-wise datasets using the life tables above as follows: for the means, we use the estimated survival probabilities, for the standard deviations, we transform the Greenwood standard error estimates by multiplying them with the square root of the sample size and for the sample sizes, we use the same values as were specified for the gestate recruitment curves (i.e., \(157\) patients per arm per stage). For simplification, no dropouts are assumed. The first stage (continuous) dataset is therefore:
# Landmark at which the analysis is to be performed
lm <- 7
# first stage dataset
dataset_1 <- getDataset(
# accessing the survival probability values at the timepoint closest to the desired one
means1 = c(lt_t1_1$surv[which(abs(lt_t1_1$time - lm) == min(abs(lt_t1_1$time - lm)))]),
means2 = c(lt_t2_1$surv[which(abs(lt_t2_1$time - lm) == min(abs(lt_t2_1$time - lm)))]),
means3 = c(lt_c_1$surv[which(abs(lt_c_1$time - lm) == min(abs(lt_c_1$time - lm)))]),
stDevs1 = c(lt_t1_1$std.err[which(abs(lt_t1_1$time - lm) ==
min(abs(lt_t1_1$time - lm)))] * sqrt(n)),
stDevs2 = c(lt_t2_1$std.err[which(abs(lt_t2_1$time - lm) ==
min(abs(lt_t2_1$time - lm)))] * sqrt(n)),
stDevs3 = c(lt_c_1$std.err[which(abs(lt_c_1$time - lm) ==
min(abs(lt_c_1$time - lm)))] * sqrt(n)),
n1 = c(n),
n2 = c(n),
n3 = c(n)
)
dataset_1 |> summary()Dataset of multi-arm means
The dataset contains the sample sizes, means, and standard deviations of two treatment groups and one control group.
| Stage | 1 | 1 | 1 |
|---|---|---|---|
| Group | 1 | 2 | 3 |
| Sample size | 157 | 157 | 157 |
| Mean | 0.734 | 0.761 | 0.543 |
| Standard deviation | 0.657 | 0.661 | 0.811 |
The explicit estimates can be seen using the summary()-command. The highest index (\(=3\)) represents the control group.
Note that the time variable in the life table data has not been structured into intervals of equal length and therefore, it is possible that the stage-wise life tables do not have entries at the exact timing of the intended landmark analyses. For this reason, the survival probabilities and standard errors are extracted from the entries for which the time variable is closest to the intended landmark time point.
Analysis of the data according to the inverse normal design derived earlier in this vignette can then be done by:
result_1 <- getAnalysisResults(
dataInput = dataset_1,
design = d_IN,
intersectionTest = "Simes",
normalApproximation = TRUE,
varianceOption = "pairwisePooled"
)result_1 |> summary()Multi-arm analysis results for a continuous endpoint (2 active arms vs. control)
Sequential analysis with 3 looks (inverse normal combination test design), one-sided overall significance level 2.5%. The results were calculated using a multi-arm t-test, Simes intersection test, normal approximation test, pairwise pooled variances option. H0: mu(i) - mu(control) = 0 against H1: mu(i) - mu(control) > 0.
| Stage | 1 | 2 | 3 |
|---|---|---|---|
| Fixed weight | 0.577 | 0.577 | 0.577 |
| Cumulative alpha spent | 0.0001 | 0.0060 | 0.0250 |
| Stage levels (one-sided) | 0.0001 | 0.0060 | 0.0231 |
| Efficacy boundary (z-value scale) | 3.710 | 2.511 | 1.993 |
| Futility boundary (z-value scale) | 0.149 | 0.414 | |
| Cumulative effect size (1) | 0.191 | ||
| Cumulative effect size (2) | 0.217 | ||
| Cumulative (pooled) standard deviation | 0.714 | ||
| Stage-wise test statistic (1) | 2.287 | ||
| Stage-wise test statistic (2) | 2.601 | ||
| Stage-wise p-value (1) | 0.0111 | ||
| Stage-wise p-value (2) | 0.0046 | ||
| Adjusted stage-wise p-value (1, 2) | 0.0093 | ||
| Adjusted stage-wise p-value (1) | 0.0111 | ||
| Adjusted stage-wise p-value (2) | 0.0046 | ||
| Overall adjusted test statistic (1, 2) | 2.354 | ||
| Overall adjusted test statistic (1) | 2.287 | ||
| Overall adjusted test statistic (2) | 2.601 | ||
| Test action: reject (1) | FALSE | ||
| Test action: reject (2) | FALSE | ||
| Conditional rejection probability (1) | 0.2359 | ||
| Conditional rejection probability (2) | 0.2530 | ||
| 95% repeated confidence interval (1) | [-0.133; 0.514] | ||
| 95% repeated confidence interval (2) | [-0.107; 0.542] | ||
| Repeated p-value (1) | 0.1425 | ||
| Repeated p-value (2) | 0.1331 |
Legend:
- (i): results of treatment arm i vs. control arm
- (i, j, …): comparison of treatment arms ‘i, j, …’ vs. control arm
Since the overall adjusted test statistic for the intersection hypotheses (\(=2.354\)) does not exceed the first stage critical value of \(3.710\), the intersection hypothesis of having no effect in either one of the treatment groups cannot be rejected, and, e.g., the repeated p-values lead to the same conclusion. Furthermore, since all of the applicable test statistics do not fall below the futility bound of \(0.149\), the study proceeds to the second stage with both active arms and the dataset is created in the same way as above:
dataset_2 <- getDataset(
means1 = c(
lt_t1_1$surv[which(abs(lt_t1_1$time - lm) == min(abs(lt_t1_1$time - lm)))],
lt_t1_2$surv[which(abs(lt_t1_2$time - lm) == min(abs(lt_t1_2$time - lm)))]
),
means2 = c(
lt_t2_1$surv[which(abs(lt_t2_1$time - lm) == min(abs(lt_t2_1$time - lm)))],
lt_t2_2$surv[which(abs(lt_t2_2$time - lm) == min(abs(lt_t2_2$time - lm)))]
),
means3 = c(
lt_c_1$surv[which(abs(lt_c_1$time - lm) == min(abs(lt_c_1$time - lm)))],
lt_c_2$surv[which(abs(lt_c_2$time - lm) == min(abs(lt_c_2$time - lm)))]
),
stDevs1 = c(
lt_t1_1$std.err[which(abs(lt_t1_1$time - lm) ==
min(abs(lt_t1_1$time - lm)))] * sqrt(n),
lt_t1_2$std.err[
which(abs(lt_t1_2$time - lm) == min(abs(lt_t1_2$time - lm)))
] * sqrt(n)
),
stDevs2 = c(
lt_t2_1$std.err[which(abs(lt_t2_1$time - lm) ==
min(abs(lt_t2_1$time - lm)))] * sqrt(n),
lt_t2_2$std.err[which(abs(lt_t2_2$time - lm) ==
min(abs(lt_t2_2$time - lm)))] * sqrt(n)
),
stDevs3 = c(
lt_c_1$std.err[which(abs(lt_c_1$time - lm) == min(abs(lt_c_1$time - lm)))]
* sqrt(n),
lt_c_2$std.err[which(abs(lt_c_2$time - lm) == min(abs(lt_c_2$time - lm)))]
* sqrt(n)
),
n1 = c(n, n),
n2 = c(n, n),
n3 = c(n, n)
)
result_2 <- getAnalysisResults(
dataInput = dataset_2, design = d_IN,
intersectionTest = "Simes", normalApproximation = TRUE,
varianceOption = "pairwisePooled"
)result_2 |> summary()Multi-arm analysis results for a continuous endpoint (2 active arms vs. control)
Sequential analysis with 3 looks (inverse normal combination test design), one-sided overall significance level 2.5%. The results were calculated using a multi-arm t-test, Simes intersection test, normal approximation test, pairwise pooled variances option. H0: mu(i) - mu(control) = 0 against H1: mu(i) - mu(control) > 0.
| Stage | 1 | 2 | 3 |
|---|---|---|---|
| Fixed weight | 0.577 | 0.577 | 0.577 |
| Cumulative alpha spent | 0.0001 | 0.0060 | 0.0250 |
| Stage levels (one-sided) | 0.0001 | 0.0060 | 0.0231 |
| Efficacy boundary (z-value scale) | 3.710 | 2.511 | 1.993 |
| Futility boundary (z-value scale) | 0.149 | 0.414 | |
| Cumulative effect size (1) | 0.191 | 0.226 | |
| Cumulative effect size (2) | 0.217 | 0.329 | |
| Cumulative (pooled) standard deviation | 0.714 | 0.931 | |
| Stage-wise test statistic (1) | 2.287 | 1.769 | |
| Stage-wise test statistic (2) | 2.601 | 3.512 | |
| Stage-wise p-value (1) | 0.0111 | 0.0384 | |
| Stage-wise p-value (2) | 0.0046 | 0.0002 | |
| Adjusted stage-wise p-value (1, 2) | 0.0093 | 0.0004 | |
| Adjusted stage-wise p-value (1) | 0.0111 | 0.0384 | |
| Adjusted stage-wise p-value (2) | 0.0046 | 0.0002 | |
| Overall adjusted test statistic (1, 2) | 2.354 | 4.015 | |
| Overall adjusted test statistic (1) | 2.287 | 2.868 | |
| Overall adjusted test statistic (2) | 2.601 | 4.323 | |
| Test action: reject (1) | FALSE | TRUE | |
| Test action: reject (2) | FALSE | TRUE | |
| Conditional rejection probability (1) | 0.2359 | 0.7272 | |
| Conditional rejection probability (2) | 0.2530 | 0.9870 | |
| 95% repeated confidence interval (1) | [-0.133; 0.514] | [-0.005; 0.441] | |
| 95% repeated confidence interval (2) | [-0.107; 0.542] | [0.096; 0.527] | |
| Repeated p-value (1) | 0.1425 | 0.0119 | |
| Repeated p-value (2) | 0.1331 | 0.0007 |
Legend:
- (i): results of treatment arm i vs. control arm
- (i, j, …): comparison of treatment arms ‘i, j, …’ vs. control arm
Again, referring to the overall adjusted test statistics, since \(4.015 > 2.511\), the global intersection hypothesis can be rejected in the second stage. Subsequently, the hypothesis of no effect in the first treatment group can be rejected because \(2.868 > 2.511\) and the hypothesis of no effect in the second treatment group can be rejected due to \(4.323\) exceeding the applicable critical value. Another testing approach would be to compare repeated p-values listed at the end of the output (\(0.0119, \, 0.0007\)) to the full significance level \(\alpha=0.025\).
As mentioned in the context of the simulations in chapter 5, study success can only be concluded when both treatment arms have been tested significantly against control. As this appears to be the case here, the study would have successfully finished at the second stage.
Closing remarks
This vignette demonstrated how to implement a MAMS study design in case the futility bounds are only known on the treatment effect scale. In addition, how to perform MAMS simulations with special regards to treatment arm selection as well as how to analyze generic data using the binary module or perform landmark analysis with the support of gestate was demonstrated. Especially considering the final chapter about the landmark analysis, further investigations on how the results and characteristics such as \(\alpha\)-level control and/or the power behave could be done in subsequent research.
System: rpact 4.4.0.9305, R version 4.6.0 (2026-04-24), platform: x86_64-pc-linux-gnu
To cite R in publications use:
R Core Team (2026). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. doi:10.32614/R.manuals https://doi.org/10.32614/R.manuals. https://www.R-project.org/.
To cite package ‘rpact’ in publications use:
Wassmer G, Pahlke F (2026). rpact: Confirmatory Adaptive Clinical Trial Design and Analysis. R package version 4.4.0.9305. doi:10.32614/CRAN.package.rpact
Wassmer G, Brannath W (2025). Group Sequential and Confirmatory Adaptive Designs in Clinical Trials, 2nd edition. Springer, Cham, Switzerland. ISBN 978-3-031-89668-2. doi:10.1007/978-3-031-89669-9 https://doi.org/10.1007/978-3-031-89669-9.